Фармаконадзор : Побочные реакции лекарств

Памятник жертвам талидомида в Лондоне (установлен временно и будет перевозиться из города в город). Талидомид — лекарство, которое давали беременным в 1962 году, чтобы сгладить эффект от токсикоза. Результат: несколько тысяч врожденных уродств конечностей у новорожденных детей.

Скульптуру Марку Куинну позировала реально существующая женщина Элисон Леппер, которая в момент создания памятника была беременна. Ее ребенок вырос здоровым. Талидомид до сих пор разрешен в лечебной практике как противораковое средство.
Побочные реакции (ПР) лекарственных средств (ЛС) существенно ухудшают качество жизни, вызывают необходимость госпитализации или увеличивают продолжительность пребывания на больничной койке, могут быть причиной непоправимого вреда для здоровья и даже смерти (Pirmohamed M. et al., 2004). Сбор и анализ такой информации необходим для принятия решений относительно дальнейшего пребывания лекарственного средства на фармацевтическом рынке. Надзор за безопасностью ЛС (фармаконадзор) — сложная задача, требующая вовлечения всех сторон, участвующих в обращении ЛС и их применении. Однако врачи недостаточно мотивированы для передачи информации о ПР в регуляторные органы и производителям, а пациенты зачастую получают неадекватную и недостаточную информацию о ПР вообще и у себя в частности, и их сообщения редко бывают достаточно информативными. Между тем, количество новых препаратов с каждым годом увеличивается, их применяет все большее количество людей, появляются ЛС, полученные с применением совершенно новых технологий, в силу чего их эффекты труднопрогнозируемы. В связи с этим фармаконадзор — быстро меняющаяся и прогрессивно развивающаяся система.
Словосочетание «талидомидовая трагедия» постепенно теряет тот ужасающий смысл, который она имела для современников в начале 60-х годов, когда на свет появились дети с врожденными деформациями конечностей. Эти события заставили мировую общественность содрогнуться и впервые серьезно задуматься о необходимости контроля за безопасностью ЛС. Возникла необходимость в системе, которая бы позволила накапливать информацию о побочных реакциях, критически ее оценивать, предоставляла возможность систематизировать сообщения, производить статистический анализ полученной информации и делать обоснованные выводы.
Попробуем представить основные инициативы по фармаконадзору в хронологическом порядке. С начала ХХ века на фоне роста количества ЛС на рынке накапливались сведения о неправильно маркированных, неэффективных или опасных продуктах. К серьезным изменениям в американском законодательстве привел случай в 1937 г., когда свыше 100 детей погибли после приема препарата из группы сульфаниламидов в форме сиропа «Эликсир сульфаниламида» (Elixir sulfanilamide), в котором в качестве вспомогательного вещества содержался диэтиленгликоль — высокотоксичное вещество, применяемое в качестве антифриза (www.fda.gov). С тех пор накопилось немало примеров случаев серьезных ПР, но переломным для всего мирового сообщества, особенно для европейских стран, стал 1961 г., когда только в Западной Германии после приема беременными препарата талидомида родились 10 000 детей с фокомегалией (гипопластическими или апластическими деформациями конечностей).
В октябре 1962 г. в США президент Дж. Кеннеди утвердил поправку к Федеральному закону о пищевых продуктах и лекарственных средствах (Federal Food and Drugs Act), вводящую требование доказательства и эффективности, и безопасности препарата перед выдачей разрешения на его маркетинг. Во исполнение изменений в законодательстве Отдел новых препаратов (Division of New Drugs) Управления по контролю за пищевыми продуктами и лекарственными средствами США (Food and Drug Administration — FDA) был разделен на пять подразделений, одно из которых — по надзору за новыми лекарствами (New Drug Surveillance), стало заниматься сбором сведений о ПР.
В 1968 г. Всемирная организация здравоохранения (ВОЗ) учредила Международную программу по мониторингу лекарственных средств (Drug Monitoring Programme) (далее — Программа). С 1978 г. работу по Программе начинает выполнять Центр Сотрудничества по Международному Мониторингу Лекарственных препаратов (Collaborating Centre for International Drug Monitoring), иначе его называют Уппсаловским мониторинговым центром (Uppsala Monitoring Centre) (штаб-квартира в г. Уппсала, Швеция). С 1968 г. в Программе ВОЗ участвуют Австралия, Канада, Дания, Франция, Германия, Нидерланды, Новая Зеландия, Швеция, Ирландия, Великобритания и США. Россия, Эстония и Индия присоединились в 1998 г., Украина — в 2002 г., Беларусь, Узбекистан и Непал — в 2006 г. По состоянию на июль 2006 г. в Программе участвует 81 страна. Кроме того, 17 стран (в том числе Грузия, Казахстан, Пакистан) считаются «ассоциированными членами», проводя работу по достижению совместимости национального и международного формата сообщений о ПР.
Европейское Сообщество было пионером в гармонизации регуляторных требований между странами, начав создание единого фармацевтического рынка стран — членов ЕС в 1980-х гг. Последовавшие переговорные процессы с участием США и Японии привели к созданию Международной конференции по гармонизации технических требований к регистрации лекарственных средств для человека, производства и продажи ЛС (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use — ICH), годом рождения которой считается 1990. В рамках ICH сегодня создаются стандартизированные форматы сообщений о ПР, периодические отчеты и руководства по управлению рисками. Большое внимание уделяется проблемам имплементации и аудиту этих форм деятельности.
С конца 2001 г. в ЕС действует EudraVigilance — сеть обработки данных и система управления сбором и оценкой информации о вероятных ПР лекарственного средства во время его разработки и после получения разрешения на маркетинг в ЕС.
СИТУАЦИЯ НА ФАРМРЫНКЕ ТРЕБУЕТ АКТИВИЗАЦИИ РАБОТЫ
В последнее время на фармацевтическом рынке появилось много новых тенденций, которые уже сейчас меняют ситуацию с безопасностью ЛС. Об этом в «Берлинской декларации по фармаконадзору» пишет Международное общество лекарственных бюллетеней (International Society of Drug Bulletins — ISDB) — всемирная информационная сеть, «финансово и интеллектуально независимая от фармацевтической отрасли» (http://66.71.191.169/isdbweb/pag/summary.php). Организация основана в 1986 г. при поддержке Европейского регионального бюро ВОЗ.
В декларации отмечается, что в последние десятилетия фармакотерапия применяется все шире, нередко для «улучшения качества жизни», а не из клинических соображений. Помимо терапевтической пользы это неизбежно влечет риск ПР. Препараты выводятся на рынок и маркетируются во многих странах одновременно. Такая глобализация деятельности фармацевтических компаний ведет к тому, что новый препарат начинают применять у многих людей сразу, что несоизмеримо больше количества участников премаркетинговых клинических испытаний. Растет рынок безрецептурных препаратов, шире используется приобретение медикаментов через интернет и без участия фармацевтов, распространяется альтернативная медицина. Контрафактная продукция находит «дорожку» даже на рынки развитых стран. Велико финансовое бремя ПР: вызванные ими финансовые затраты составляют 7–18 млн евро на 1 млн европейцев (Classen D.C., 1997). Эти и другие явления вызывают обеспокоенность в связи с повышением риска ПР, что вызывает необходимость повышения эффективности и качества работы по фармаконадзору.
В США СООБЩАТЬ О ПОБОЧНЫХ РЕАКЦИЯХ ОБЯЗАНЫ ВСЕ ОПЕРАТОРЫ РЫНКА
В Украине нормативную базу фармакологического надзора стремятся гармонизировать с руководствами ICH и нормами ЕС. Но опыт FDA — одного из старейших регуляторных органов в области фармации в мире, без сомнения, тоже интересен. Тем более, что законодательные положения ЕС — это документ для имплементации в национальные законы и нормы, так что их анализ не дает полного представления о сложившемся регуляторном окружении в стране. Например, поскольку отношение к сохранению конфиденциальности информации в различных европейских странах отличается, в руководствах и руководящих принципах ЕС очень мало конкретных указаний на этот счет (см. «Еженедельник АПТЕКА» № 33 (554) от 28.08.2006 г.).
Подразделением Центра по оценке и исследованиям лекарственных средств (Center for Drug Evaluation and Research — CDER) FDA, осуществляющим оценку профилей безопасности ЛС, обмен информацией со специалистами и пациентами, является Служба безопасности ЛС (Office of Drug Safety — ODS). С 2001 г. в составе Консультативного комитета по фармацевтической науке (Advisory Committee for Pharmaceutical Science) действует новый консультативный субкомитет по безопасности ЛС и управлению рисками. Постмаркетинговый надзор за безопасностью ЛС выполняется при помощи компьютеризированной базы данных «Система сообщений о ПР» (Adverse Event Reporting System — AERS). В нее поступают постмаркетинговые 15-дневные экстренные сообщения (postmarketing 15-day «alert reports»), регулярно обновляемые отчеты по безопасности (Periodic Safety Update Reports — PSUR) и сообщения от специалистов здравоохранения и пациентов (посредством программы MedWatch).
Перечисленные документы — это и есть те основные три вида сообщений, которые используются повсеместно: добровольные от специалистов здравоохранения и пациентов, обязательные экстренные от производителей и исследователей и периодически подаваемые — PSUR от производителей.
В США обязанность подавать экстренные отчеты возложена не только на владельца разрешения на маркетинг. В соответствии со статьей 310.305 Кодекса федеральных правил (Code of Federal Regulations — CFR) каждая организация, чье название указано в инструкции препарата (производитель, упаковщик или дистрибьютор), обязана сообщать FDA о каждом случае серьезных непредвиденных ПР, но не позднее, чем спустя 15 дней после получения такой информации. Для избежания ненужного дублирования этих сведений в FDA их может подавать владелец разрешения на маркетинг, к которому соответствующие сведения должны поступить не позднее, чем 5 дней спустя после их получения другим субъектом. Обо всех остальных ПР (вместе с анализом срочных сообщений) владелец разрешения на маркетинг должен сообщать FDA при помощи PSUR раз в квартал на протяжении 3 лет после даты получения разрешения на маркетинг, а затем с годичными интервалами.
Сотрудники системы здравоохранения и потребители могут сообщать о вероятных ПР посредством системы MedWatch, где возможны коммуникации при помощи интернета, электронной почты, факса и телефонных переговоров («Еженедельник АПТЕКА» № 31 (502) от 15.08.2005 г.). Действует и программа MedWatch для производителя, которая позволяет владельцу разрешения на маркетинг получать отчеты о серьезных ПР, которые добровольно предоставлены в FDA. Следует отметить, что форматы отчетов гармонизированы с нормами ICH и в PSUR используется стандартизированная международная терминология (Medical Dictionary for Regulatory Activities — MedDRA) (см. ниже).
Интересно, что Форма FDA 3500 — Добровольный отчет (Voluntary Reporting) по структуре похожа на отечественную Форму № 137/у (карта-сообщение), утвержденную Приказом МЗ Украины от 16.07.2001 г. № 292 (опубликован в «Еженедельнике АПТЕКА», № 30 (301) от 06.08.2001 г.). Последняя предусматривает несколько более подробное предоставление информации, чем упомянутая форма FDA. Так, требуются все данные (клинические, лабораторные, рентгенологические и др.) до назначения подозреваемого ЛС или до возникновения ПР/побочного действия (ПД), а также после выявления ПР/ПД, указываются даты обследований и анализов. Необходимо также описывать меры, предпринятые для коррекции проявлений ПР/ПД. Наконец, карта-сообщение предоставляется врачом главному врачу лечебно-профилактического учреждения, тогда как Форма FDA 3500 поступает непосредственно в агентство, которому не позволено разглашать сведения о сообщившем (см. «Еженедельник АПТЕКА» № 33 (554) от 28.08.2006 г.). Если того не желает отправитель сообщения и делает соответствующую пометку в заполненной форме FDA 3500, его не сможет идентифицировать и владелец разрешения на маркетинг подозреваемого ЛС, пользующийся программой MedWatch для производителя.
Мало различаются форма FDA 3500A — Обязательный отчет (Mandatory reporting) и отечественная форма «Карта сообщений о любых подозреваемых серьезных и/или непредвиденных ПР/ПД ЛС». В американском варианте карту заполняет исследователь, импортер, дистрибьютор или производитель, в украинском — производитель/владелец регистрационного свидетельства или их уполномоченный представитель.
В 2004 фискальном году в ODS поступило свыше 426 тыс. экстренных сообщений и PSUR. Приоритетной задачей остается расширение применения электронного формата подачи, что позволяет ускорить получение и обработку сообщений и снизить расходы, связанные с пересылкой. С января 2005 г. ежеквартальные выдержки из AERS доступны для загрузки
(www.fda.gov/cder/aers/extract.htm).
С 2002 г. ODS выполняет еще одну важную функцию: изучение и оценку планов управления рисками и определение необходимости постмаркетинговых обсервационных исследований. В период рассмотрения заявки на получение разрешения на маркетинг ЛС, в том числе биологического происхождения, ODS участвует в составлении соответствующих программ и на протяжении 2–3 лет после получения разрешения на маркетинг оценивает выполнение этих планов. В 2004 финансовом году, о чем сообщается в ежегодном отчете, ODS участвовала в составлении 23 планов управления рисками (www.fda.gov).
НОВОЕ ФАРМАЦЕВТИЧЕСКОЕ ЗАКОНОДАТЕЛЬСТВО ЕС И ПРАВИЛА ФАРМАКОНАДЗОРА
О регулировании системы фармаконадзора в ЕС мы уже сообщали («Еженедельник АПТЕКА» № 5 (376) от 10.02.2003 г.), издана книга «Фармацевтический сектор, фармаконадзор за лекарственными препаратами для человека» («МОРИОН», 2003). В новом фармацевтическом законодательстве, которое призвано дополнить и расширить положения Директивы 2001/83/ЕС (дополненной Директивами 2002/98/ЕС, 2004/24/ЕС, 2004/27/ЕС) и Директивы 2001/82/ЕС (дополненной Директивой 2004/28/ЕС), правила осуществления фармаконадзора получили дальнейшее развитие. Так, согласно положениям Регламента (ЕС) № 726/2004, вступившим в силу весной 2004 г., меняется периодичность подачи PSUR. Если условиями выдачи разрешения на маркетинг в Содружестве не предусмотрено иначе, указывается в статье 24 Регламента, PSUR подаются в Европейское агентство по лекарственным средствам (European Medicines Agency — EMEA) и стране-участнице немедленно по требованию или по меньшей мере 1 раз в 6 мес после получения разрешения на маркетинг до выведения препарата на рынок. Также немедленно по требованию или по меньшей мере 1 раз в 6 мес PSUR должны подаваться в течение первых двух лет после выведения на рынок Содружества, затем — ежегодно в последующие 2 года. Затем PSUR следует подавать с 3-летними интервалами или немедленно по требованию. Эти новые правила применяются по отношению к препаратам, одобренным по централизованной процедуре после 20 ноября 2005 г. Если разрешение на маркетинг выдано ранее этой даты, новая периодичность подачи PSUR должна применяться после очередного обновления разрешения на маркетинг, производящегося 1 раз в 5 лет на основании повторной оценки EMEA соотношения польза/эффективность. PSUR следует сопровождать научным анализом, в частности, что касается соотношения польза/риск ЛС.
Владелец разрешения на маркетинг и уполномоченный орган страны-участницы должны обеспечить предоставление вниманию EMEA всей релевантной информации относительно вероятных побочных реакций препарата, получившего одобрение в соответствии с Регламентом (ЕС) № 726/2004. Владелец разрешения на маркетинг должен обеспечить информирование страны-члена обо всех вероятных ПР, которые произошли на его территории, не позднее, чем через 15 дней после получения таких сведений от специалиста здравоохранения. При этом он может не доносить до широкой общественности информацию относительно выявленных при фармаконадзоре проблем. Страны-участницы должны принимать необходимые меры, обеспечивающие, чтобы владелец разрешения на маркетинг, не выполнивший указанные обязательства, подвергался эффективному, пропорциональному и действенному взысканию. Такие положения содержатся в главе 3 «Фармаконадзор» Регламента (ЕС) № 726/2004.
Более подробная информация о правилах фармаконадзора в ЕС содержится в руководстве «Фармаконадзор. Лекарственные препараты для человека и для применения в ветеринарии» (Volume 9. Pharmacovigilance. Medicinal Products for Human and Veterinary Use) (его перевод представлен в вышеупомянутой книге). К настоящему времени уже закончилось обсуждение новой версии руководства по фармаконадзору, которая должна заменить действующую редакцию EUDRALEX том 9 — «Фармаконадзор». Новый документ более подробный и содержит дополнительные разделы, например, по коммуникациям между владельцем разрешения на маркетинг и уполномоченным органом, по фармаконадзору, специфичному для отдельных популяций или продуктов.
Новым для европейского фармацевтического законодательства являются системы управления рисками. Только в 2005 г. введено требование подачи такой информации вместе с заявкой на получение разрешения на маркетинг. Предпосылкой стало понимание того, что планирование деятельности по фармаконадзору может быть улучшено, если она будет более прочно базироваться на специфических для того или иного продукта проблемах, выявленных на разных стадиях жизненного цикла препарата. Системам управления рисками посвящено отдельное руководство, вступившее в силу в ноябре 2005 г. Оно будет включено в виде отдельной главы в EUDRALEX том 9 («Guideline on Risk Management Systems for Medicinal Products for Human Use»). Необходимость предоставления плана управления рисками (EU Risk Management Plan — EU-RMP) может возникнуть в любой фазе жизненного цикла продукта, указывается в руководстве. В частности, EU-RMP следует подавать:
- с заявкой на получение разрешения на маркетинг
- любого препарата, содержащего новую активную субстанцию;
- аналогичного биологического ЛС;
- генерического ЛС, если из соображений безопасности требуется дополнительная деятельность по минимизации риска, который был выявлен у референтного препарата;
- если дополнительная заявка содержит значительные изменения к уже существующей;
- по требованию уполномоченного органа.
В регистрационном досье план EU-RMP, снабженный соответствующими документами, такими, как протоколы исследований, помещают в Модуль 1. Обобщение важных идентифицированных, потенциальных рисков и важной недостающей информации называют спецификацией по безопасности (Safety Specification). В руководстве представлена рекомендуемая структура этих спецификаций. В нее включены следующие элементы:
- неклинические данные (токсикологические, общефармакологические, относительно лекарственных взаимодействий), значение которых не полностью прояснено в ходе клинических испытаний;
- клинические:
- ограничения существующих данных о безопасности по причине недостаточного количества исследованных, критериев включения/исключения и т.п.);
- постмаркетинговые данные о количестве пациентов, принимавших данное ЛС, предпочтительно на основании результатов исследований рынка;
- неисследованные в премаркетинговой стадии популяции пациентов;
- постмаркетинговый опыт (чем реальное применение препарата отличалось от предполагаемого, в том числе согласно утвержденным показаниям и противопоказаниям);
- ПР (выявленные и потенциальные) — приведен рекомендуемый формат подачи этой информации;
- выявленные и потенциальные взаимодействия с другими ЛС и пищевыми продуктами;
- эпидемиологическая характеристика ситуации с распространенностью ситуаций, при которых показан препарат;
- эффекты, общие для препаратов фармакологического класса;
- дополнительные параметры для обсуждения (возможность передозировки, передачи инфекционных заболеваний, применения в нелегальных целях, не по показаниям, в том числе в педиатрии).
Если применение продукта связано с важными факторами риска, помимо рутинных мер по фармаконадзору предпринимаются дополнительные. Обе разновидности включаются в разрабатываемые владельцем разрешения на маркетинг совместно с уполномоченным органом EU-RMP. Этот план должен включать обобщение намеченных действий, состоящее из двух частей: (1) обобщение мер по каждой важной проблеме безопасности; (2) обобщение всех мер в хронологическом порядке. В дополнении к руководству собрана информация об эпидемиологических методах, используемых для постмаркетинговых исследований по безопасности ЛС.
Система обработки и управления данными «EudraVigilance», с помощью которой осуществляется фармаконадзор как в пре-, так и постмаркетинговой фазах существования ЛС, содержит два основных отчетных модуля:
- EudraVigilance Clinical Trial Module (EVCTM) — поддерживает электронное информирование о вероятных непредвиденных серьезных побочных реакциях, как требует Директива 2001/20/ЕС («Еженедельник АПТЕКА» № 48 (469) от 13.12.2004 г.);
- EudraVigilance Post-Authorisation Module (EVPM) — предназначена для спонтанных сообщений о подозреваемых ПР (Individual Case Safety Reports) после выдачи разрешения на маркетинг.
В системе EudraVigilance каждой зарегистрированной организации предоставляется специфический идентификатор (ID), что позволяет корректно идентифицировать двух уникальных партнеров в электронном обмене информацией. Больше того, система позволяет мониторировать взаимодействие с базой данных конкретных пользователей из тех или иных организаций. Поскольку идентифицированы источники поступления отчетов, можно реализовывать различный объем доступа к информации. Например, владельцы разрешений на маркетинг могут просматривать только те отчеты, которые они сами и отправили.
РОЛЬ ICH
В последние годы регуляторными органами и отраслевыми ассоциациями предпринимается большое количество инициатив, направленных на международную гармонизацию регуляторных требований. Взаимодействие и координация усилий в сфере постмаркетингового надзора считается одним из приоритетов ICH. Целью является выявление и устранение различий в содержании, терминологии и формате отчетов, частоте их предоставления. С середины 1990-х гг. FDA и ЕМЕА разработали ряд руководств и руководящих принципов на основе документов, созданных ICH и Советом международных научно-медицинских организаций (Council for International Organizations of Medical Sciences —CIOMS):
- ICH-E2A; сформулированы определения и стандарты, используемые для экстренных сообщений (экспресс-отчетах) о побочных эффектах во время клинических испытаний;
- ICH-E2В; о передаче сообщений о побочных эффектах;
- ICH-E2С; о PSUR;
- ICH-E2D; о процедурах и стандартах обращения с постмаркетинговыми данными о безопасности;
- ICH-E2D; о планировании деятельности по фармаконадзору регуляторных органов и предприятий отрасли;
- ICH M1; по медицинской терминологии (Medical Terminology — MedDRA).
НАДЛЕЖАЩАЯ ПРАКТИКА РАБОТЫ С СООБЩЕНИЯМИ
В статье, опубликованной в прошлом выпуске «Еженедельника АПТЕКА», подробно рассказано о формировании и подаче PSUR («Еженедельник АПТЕКА», № 34 (555) от 4.09.2006 г.). Представляется, что есть смысл остановиться на стандартах работы с сообщениями о побочных эффектах ЛС или надлежащей практике такой деятельности. Именно такое выражение используется в соответствующем европейском руководстве (E2D «Post-Approval Safety Data Management: Definitions and Standards for Expedited Reporting»), одобренном EMEA в ноябре 2003 г.
| Таблица 1 | |||||||||||||||||||||
| Три группы препаратов по АТС-классификации (и по 5 ЛС в каждой из них), о ПР которых сообщения в 2000–2004 гг. поступали наиболее часто из стран Северной Америки и Европы | |||||||||||||||||||||
|
|||||||||||||||||||||
| Таблица 2 | |||||||||||||||||||||
| Топ-10 ЛС, о которых сообщения из США и стран Европы поступали наиболее часто | |||||||||||||||||||||
|
Фармацевтические компании могут пользоваться следующими источниками для получения индивидуальных сообщений о случаях ПР ЛС:
1. С необязательной, не исчерпывающей фиксацией данных.
-
Спонтанные сообщения — это добровольная передача информации об одном или более ПД при медицинском применении одного или нескольких ЛС от специалиста здравоохранения или потребителя (пациент, адвокат или его близкие) компании, регуляторному органу или другим организациям (ВОЗ, региональные центры), если эта информация не получена в ходе исследований или любой другой организованной схемы сбора данных.
Подача спонтанных сообщений может быть стимулирована в некоторых ситуациях при помощи писем к специалистам здравоохранения («Dear Healthcare Professional» letters), публикаций в прессе или непосредственными просьбами медицинских представителей.
С сообщениями от потребителей необходимо обращаться как со спонтанными сообщениями, вне зависимости от наличия «медицинского подтверждения» (оно может потребоваться для передачи экстренных сообщений в регуляторные органы).
- Литературные данные. Считается, что каждый владелец разрешения на маркетинг регулярно просматривает мировую научную литературу, изучая систематические обзоры или реферативную информацию. Частота выполнения литературного поиска должна соответствовать локальным требованиям или производиться по меньшей мере каждые 2 недели. Случаи ПР, описанные в научной и медицинской литературе, включая материалы конференций и проекты рукописей, могут в форме экстренных сообщений поступать в регуляторные органы. Соответствующая форма должна заполняться на каждого идентифицированного пациента. Ссылка на публикацию, копия которой может потребоваться в дополнение к сообщению, должна считаться его источником. Необходимо поощрять литературный поиск в местных изданиях сотрудниками представительств, с последующим отправлением публикаций в отдел безопасности компаний. Если в литературе не указан производитель или торговая марка продукта, владелец разрешения на маркетинг отправляет сообщение так же, как о своем продукте, делая соответствующую пометку.
- Интернет. От владельца разрешения на маркетинг не ожидают, что он будет регулярно просматривать внешние веб-сайты в поисках информации о ПР. Но если эти сведения попали в поле его зрения, он должен рассмотреть случай и сообщить о нем. Производителю следует предусмотреть использование своего веб-сайта для сбора информации о побочных эффектах, например, предоставляя электронные формы для заполнения и сообщая необходимую контактную информацию.
- Другие источники. Если владельцу разрешения на маркетинг становится известно о случае развития ПР из немедицинских источников, например, средств массовой информации, он должен отнестись к этой информации как к спонтанному сообщению.
2. Системы с обязательной фиксацией данных, например, клинические испытания, регистры, программы поддержки пациентов, программы управления рисками и т.п. Сообщения о ПР, полученные из этих источников, не считаются спонтанными.
3. Взаимовыгодные соглашения о сотрудничестве по сбору информации о ПР между компаниями, которые совместно маркетируют тот или иной продукт. При этом ответственность за передачу информации о ПР в регуляторные органы несет владелец разрешения на маркетинг.
4. Регуляторные органы. Каждое сообщение о ПР ЛС, полученное от зарубежного регуляторного органа, подлежит экстренной пересылке владельцем разрешения на маркетинг другим регуляторным органам.
Каким минимальным критериям информативности должно соответствовать сообщение? Рекомендуется, чтобы было собрано как можно больше информации о случае, но для передачи в регуляторный орган сообщение должно содержать как минимум следующие элементы: идентифицируемые сообщивший и пациент, выявленная ПР и подозреваемый продукт. Нехватка одного из четырех компонентов позволяет считать описание случая неполным. Какие временные рамки для сообщения установлены? Экстренное сообщение о серьезной и неожиданной ПР должно подаваться как можно скорее, но не позднее, чем через 15 дней после получения. Отсчет времени начинается с того момента, когда любой представитель владельца разрешения на маркетинг получает сообщение о случае, удовлетворяющем минимальным критериям, указанным выше. Если сообщение неполное, началом 15-дневного периода считается получение дополнительных сведений. Сообщения о ПР, не расцениваемых как серьезные, обычно включают в PSUR. Какие параметры идентифицируют пациента и сообщившего? В качестве идентификатора автоматически выступает один или несколько из следующих параметров: возраст (или возрастная категория), пол, инициалы, дата рождения, имя или идентификационный номер. Если сообщение получено из вторых рук, следует прилагать все возможные усилия для установления существования сообщившего и пациента. Что следует делать после получения сообщения? Необходимо тщательно изучить случай с медицинской точки зрения. Предпочтительно получать информацию непосредственно от специалистов, оказывавших медицинскую помощь пациенту. Как оптимизировать последующее наблюдение за ПР ЛС? Сообщение о ПР при первом получении обычно недостаточно полное. Планируя изучение случаев, необходимо распределять их по приоритетности на (1) серьезные непредвиденные; (2) серьезные, но предвиденные; (3) несерьезные ПР. В какой форме сообщать? Выбор формата не является решающим, важно, чтобы информативность была максимальной. Рекомендуется пользоваться Медицинским словарем для регуляторной деятельности (Medical Dictionary for Regulatory Activities — MedDRA).
VIGIBASE — СУММАРНАЯ БАЗА ДАННЫХ СТРАН-УЧАСТНИЦ
В рамках уже упоминавшейся Международной программы по мониторингу лекарственных средств собирается уникальная база данных о ПР ЛС (Vigibase). С 1968 г. до середины 2006 г. в нее поступило около 3,7 млн сообщений из стран, участвующих в этой Программе. Сегодня Vigibase представляет собой компьютеризированную систему фармаконадзора с ежедневно обновляемой, структуризованной информацией, что упрощает поиск и анализ. Vigibase ведется параллельно с базами национальных центров фармаконадзора и пополняется из этих источников при помощи программного обеспечения «Vigibase Online». При этом информация не редактируется: это выполняют национальные центры.
Каких правил придерживается ВОЗ в сохранении конфиденциальности информации? Поскольку разные страны по-разному поступают в отношении таких сведений, и некоторые (США, например) позволяют любому члену общества получать информацию о случаях ПР (при этом изымаются данные, позволяющие идентифицировать сообщившего или пациента), а другие допускают использование таких сведений только регуляторными органами, ВОЗ придерживается компромиссного варианта. Через интернет базой может пользоваться каждый, положительно ответитивший на вопрос о наличии медицинского или фармацевтического образования. Кто имеет доступ к Vigibase? Открытый доступ к ней могут иметь все национальные центры, участвующие в Программе. В настоящее время онлайновым доступом пользуются около 60 из 81 стран-участниц. В каждой из стран бесплатное пользование может предоставляться не более чем 3 пользователям. Пользователи вне национальных центров должны оплачивать предоставление информации. Подборка отчетов (за год или по стране) обойдется более чем в 1 тыс. дол. США.
Существуют и другие базы данных, доступные на коммерческой основе. Например, корпорация «Thomson» — один из ведущих в мире провайдеров бизнес-информации для профессионалов, поддерживает базу данных о случаях ПР в США DIOGENES®. Информация поступает в нее из базы AERS FDA. С 1969 г. по январь 2006 г. в DIOGENES® накоплено свыше 3 млн сообщений о случаях ПР.
|
||||||||||
|
||||||||||
| Таблица 3 | ||||||||||
| Топ-5 фармакологических групп по частоте зарегистрированных ПД в Украине в 2005 г. (по данным ГФЦ МЗ Украины) |
||||||||||
|
КТО, ГДЕ И КАК РАБОТАЕТ СО СПОНТАННЫМИ СООБЩЕНИЯМИ?
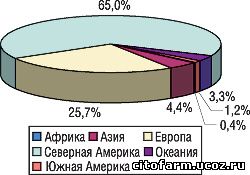
Так называется отчет по данным Vigibase за 2000–2004 гг., опубликованный на сайте Уппсаловского центра. За 5-летний исследуемый период в базу поступило свыше 217 тыс. сообщений из 73 стран. По странам и континентам они распределяются следующим образом: 62% — из США, 25,7% — из Европы (31% от этого количества — из Великобритании). Страны Азии, Океании, Южной Америки и Африки вместе обеспечили поступление менее 10% спонтанных сообщений (рис. 1).
Поэтому в Северной Америке на 1 млн жителей за год приходится 486,6, Океании — 319,5, Европе — 111,6, Южной Америке — 6,2, Африке — 3,9, Азии — 3,2 спонтанных сообщения.
Как видно из табл. 1, вакцины, антидепрессанты и противоревматические средства лидируют по количеству спонтанных сообщений из стран Европы и США, откуда о ПР сообщают намного чаще, чем из других стран-участниц. По данным из стран Океании лидируют противовоспалительные и противоревматические средства, бета-лактамные антибиотики и антипсихотические средства. Из стран Азии, Африки и Южной Америки, где сравнительно редко передают информацию о ПР, приходят сообщения, в которых в качестве подозреваемых групп ЛС первые места занимают бета-лактамные антибиотики, противовоспалительные и противоревматические средства, сульфаниламиды и триметоприм, а также антидепрессанты. Топ-10 ЛС, спонтанные сообщения о ПР при применении которых из США и Европы получали наиболее часто, показан в табл. 2.
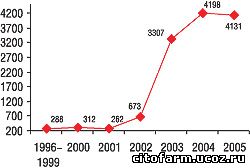
В Украине, как сообщает Государственный фармакологический центр (ГФЦ) МЗ Украины, количество полученных спонтанных сообщений в последние годы резко возросло, превысив 4 тыс., тогда как в 2000–2001 гг. их было порядка 200–300 (рис.2). Наибольшее количество ПР наблюдается при артериальной гипертензии, пневмонии, ОРЗ, пептической язве. В топ-5 фармакологических групп по частоте зарегистрированных ПД лидируют антибактериальные средства для системного применения (табл. 3). По количеству случаев лидировали следующие ЛС (в порядке убывания количества сообщений): амоксициллин, цефтриаксон, диклофенак, цефазолин, декстран, пентоксифиллин, ципрофлоксацин, эналаприл, метронидазол, ампициллин. Доля сообщений о серьезных ПР в 2003–2005 гг. составляла 10–15%. В качестве достижения последних лет нельзя не отметить рост количества сообщений о ПР, как и присоединение Украины к Программе.
ОСНОВНЫЕ ПРЕПЯТСТВИЯ
В упоминавшейся выше Берлинской декларации среди основных проблем фармаконадзора на современном этапе называют недостаточную эффективность систем сообщений о ПР, трудности с интерпретацией этих данных, недостаточную прозрачность процессов. Подчеркивается, что система спонтанных сообщений служит «костяком» фармаконадзора. Основным ее недостатком называют то, что распознать или выявить неизвестную, непредвиденную ПР специалистам здравоохранения сложно, и активность подачи спонтанных сообщений в Европе остается низкой (EMEA/CPMP Working Group with Patients Organisations. Outcome of Discussions and Proposals for Action. London, 2004). На основании сравнения данных о ПР, полученных при клинических испытаниях и путем спонтанных сообщений, предполагается, что последние обеспечивают только 2–5% уровень информирования о ПР. Специализированные центры по фармаконадзору достигают 10–20%.
Из проблем, связанных с регуляторными органами, называют недостаточную прозрачность, конфликт интересов, организационные дефекты. Как считают авторы декларации, положение ст. 26 Регламента 726/2004/ЕС — «серьезные ПР и другие данные фармаконадзора… должны становиться общедоступными, если они релеванты, после проведения оценки» — не может служить основой достаточной прозрачности деятельности ЕМЕА. Во многих странах данные, на которых основываются выводы об эффективности и безопасности, остаются неопубликованными. Информация о выявленных ПР может появляться в краткой характеристике препарата (Summary of product characteristics — SPC) спустя месяцы и годы после выявления ПР. Фармацевтические компании не в полной мере выполняют возложенную на них функцию выявлять и передавать в регуляторные органы информацию о ПР, не проводят необходимых постмаркетинговых исследований. Очень низкая активность врачей, которые не сообщают о 95–98% ПР (!).
В этой непростой ситуации нужны очень активные, консолидированные и безотлагательные меры, которые, возможно, реализуются не так полно и быстро, как хотелось бы. n
Дарья Полякова, Евгения Артеменко